|
Bios Dr. Fortenbach is an assistant professor of ophthalmology at the University of Washington, Seattle, where Dr. Hage is an ophthalmology resident, Dr. Chao a professor of ophthalmology and Dr. Bonnell a vitreoretinal surgery fellow. DISCLOSURES: The authors have no relevant relationships to disclose. |
A 60-year-old woman was referred for evaluation of macular drusen. The patient reported she was diagnosed with nonexudative age-related macular degeneration four years prior to presentation. On questioning, she reported progressive nyctalopia but denied other visual changes, including metamorphopsia.
Examination Findings
Best-corrected visual acuity of the right eye was 20/50 and left eye was 20/20. Intraocular pressures were normal in both eyes. Pupils were equal, round and reactive without a relative afferent pupillary defect.
Slit lamp biomicroscopy was unremarkable in both eyes. Dilated fundoscopic examination in both eyes revealed few macular drusen and retinal pigment epithelial mottling. There was mild vascular attenuation. There were few drusen in the periphery (Figure 1A and 1B).
Work Up
Fundus autofluorescence demonstrated hyper- and hypoautofluorescent changes throughout the posterior pole and mid-periphery (Figure 1C and 1D). Optical coherence tomography was notable for scattered sub-RPE deposits in both eyes (Figure 2). On comparison with imaging obtained four years prior to presentation to our service, there was mild blunting of the foveal contour in the right eye and progression of the sub-RPE deposits in both eyes (Figure 2B and 2D). Neither eye developed cystoid macular edema, subretinal fluid or hemorrhage.
Diagnosis
We observed that the pattern of macular drusen and fundus autofluorescence was slightly atypical for AMD. A few of the mid-peripheral autofluorescent changes had an irregular pisciform fleck-like appearance. These examination findings, coupled with the patient’s report of progressive nyctalopia, prompted consideration of a macular dystrophy. The patient was subsequently referred for genetic testing.
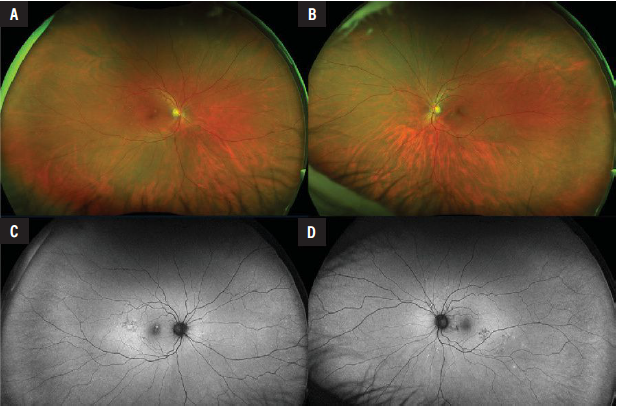 |
| Figure 1. Optos pseudocolor fundus photography of the right and left eyes demonstrating drusen around the central and temporal macula, and throughout the periphery (A, B). Vessels appear mildly attenuated. Fundus autofluorescence of the right and left eyes demonstrating hyper- and hypoautofluorescent changes throughout the macula and mid-periphery (C, D). |
Genetic testing revealed two variants in the ABCA4 gene. Mutations in this gene are associated with Stargardt disease. The first variant was a known pathogenic splice-site variant, c.4253+43G>A, that has been identified as a hypomorphic allele.1 The second variant was a likely pathogenic missense variant, c.2965G>A, p.(Val989Ile). Further genetic testing of her children revealed that her two variants were inherited separately and in trans configuration. When considering the correlation between genotype and phenotype, patients with hypomorphic and missense mutations are often found to have a milder form of Stargardt disease. Therefore, these genetic findings may explain why the patient presented with a later age of onset and a milder form of the disease than is typically seen. Ultimately, the patient was diagnosed with late-onset Stargardt disease.
Management
There’s currently no treatment available to reverse the effects of Stargardt disease. However, understanding the etiology of the patient’s macular degeneration has allowed us to recommend that the patient limit vitamin A supplementation, which is known to be associated with Stargardt disease progression. Another benefit of this diagnosis is that she may now be eligible for ongoing and upcoming clinical trials.
Discussion
Stargardt disease is the most common inherited juvenile macular dystrophy in the United States.2,3 It has an estimated prevalence of 1 in 10,000 and displays an autosomal recessive mode of inheritance.3 There is a variable age of onset among patients; however, it classically presents in childhood or early adulthood.3 Less commonly, late-onset Stargardt, as seen in our case, can develop in late adulthood and often has a better visual prognosis with sparing of the fovea in many cases.3
Stargardt disease (STGD1) is caused by mutations in the ABCA4 gene, which encodes an ATP-binding cassette transporter that‘s expressed in the outer segment of photoreceptors and in low concentrations throughout the RPE.4,5 The transporter is involved in clearing toxic byproducts of retinoid metabolism; therefore, dysfunction of the transporter leads to the accumulation of bisretinoids and A2E, which in turn cause dysfunction of the RPE, deposition of fluorescent lipofuscin and photoreceptor death.4,5
The classic presentation of Stargardt disease includes progressive loss of central vision, decreased color vision and slowed dark adaptation. Fundus examination may appear normal early in the disease course. However, over time, yellow pisciform flecks of lipofuscin deposition can be seen throughout the posterior pole and mid-periphery, as well as pigmentary mottling and macular atrophy.6
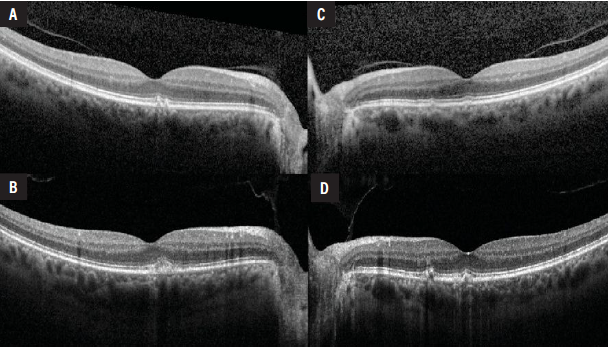 |
| Figure 2. Optical coherence tomography of the right and left eyes on initial evaluation when she was first diagnosed with AMD (A, B), compared to the first presentation to our service four years later (C, D). There are sub-RPE deposits in both eyes with evidence of progression over time. In the right eye there is mild blunting of the foveal contour (C). There is no cystoid macular edema, subretinal fluid or hemorrhage in either eye. |
Similarly, patients with AMD often experience loss of central vision, metamorphopsia, and decreased color and contrast sensitivity.7 The hallmarks of AMD include drusen and pigmentary changes throughout the macula, as well as geographic atrophy in the later stages of the disease.7 While the pathogenesis of AMD is multifactorial, dysfunction of the RPE and choroid ultimately leads to photoreceptor degeneration and central vision loss.8
Catherina H.Z. Li and colleagues recently analyzed 71 patients in the Netherlands with late-onset Stargardt disease.9 They found that the median age was 55 years; the most frequent allele was c.5603A→T (p.Asn1868Ile); and that none of the patients in the study had two severe variants.9 Most patients were found to have flecks in their fundi, foveal-sparing retinal atrophy and preserved central vision.9 Additionally, they found that 22 percent of their patients had been previously diagnosed with AMD with geographic atrophy.9 While exceedingly rare, late-onset Stargardt disease should remain on the differential for a patient with AMD, especially in cases with atypical fundoscopic findings or symptoms, such as progressive nyctalopia beyond what is observed with normal aging.
Multimodal imaging, including fundus photography, FAF and OCT, can be used in conjunction with fundoscopic examination to improve recognition of late-onset Stargardt. In general, flecks are often irregularly shaped, whereas drusen are round.9 Flecks have a less prominent yellow tint, while drusen tend to be brighter; flecks are also more hyperautofluorescent on FAF.9
Drusen are often concentrated around the macula, as opposed to flecks, which can be found throughout the posterior pole and mid-periphery.9 On OCT, drusen are defined as being under the RPE, while flecks may be situated within the photoreceptor layers.9 Additionally, foveal involvement is common in AMD, whereas late-onset Stargardt tends to spare the fovea.9
Bottom Line
While late-onset Stargardt disease and AMD may have similar clinical presentations, it’s important to distinguish between them to inform genetic and prognostic counseling, as well as to provide accurate guidance on vitamin A supplementation and eligibility for clinical trial participation. RS
REFERENCES
1. Zernant J, Lee W, Nagasaki T, et al. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb Mol Case Stud 2018;4:4:a002733.
2. Fujinami K, Zernant J, Chana RK, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 2015;122:2:326–34.
3. Tanna P, Strauss RW, Fujinami K, et al. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol 2017;101:1:25–30.
4. Lenis TL, Hu J, Ng SY, et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc Natl Acad Sci U S A 2018;115:47:E11120–E11127.
5. Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997;15:3:236–46.
6. Cremers FPM, Lee W, Collin RWJ, et al. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res 2020;79:100861.
7. Bressler SB, Do DV, Bressler NM. Age-related macular degeneration: Drusen and geographic atrophy. In: Albert DM, Miller JW, Azar DT, Blodi BA, eds. Albert and Jakobiec’s Principles and Practice of Ophthalmology. 3rd ed. Philadelphia: Saunders; 2008.
8. Fleckenstein M, Schmitz-Valckenberg S, Chakravarthy U, et al. Age-related macular degeneration: A review. JAMA 2024;331:2:147–57.
9. Li CHZ, Pas JAAH, Corradi Z, et al. Study of late-onset Stargardt type 1 disease: Characteristics, genetics, and progression. Ophthalmology 2024;131:1:87–97.